Скачать с ютуб Gaucher Disease - Pathophysiology, Diagnosis, Treatment в хорошем качестве
Gaucher Disease - Pathophysiology, Diagnosis, Treatment
3 года назад
Скачать бесплатно Gaucher Disease - Pathophysiology, Diagnosis, Treatment в качестве 4к (2к / 1080p)
У нас вы можете посмотреть бесплатно Gaucher Disease - Pathophysiology, Diagnosis, Treatment или скачать в максимальном доступном качестве, которое было загружено на ютуб. Для скачивания выберите вариант из формы ниже:
Загрузить музыку / рингтон Gaucher Disease - Pathophysiology, Diagnosis, Treatment в формате MP3:
Если кнопки скачивания не
загрузились
НАЖМИТЕ ЗДЕСЬ или обновите страницу
Если возникают проблемы со скачиванием, пожалуйста напишите в поддержку по адресу внизу
страницы.
Спасибо за использование сервиса savevideohd.ru
Gaucher Disease - Pathophysiology, Diagnosis, Treatment
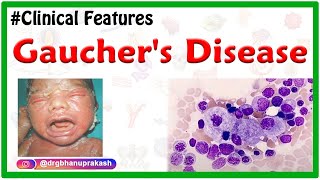
Gaucher disease is a lipid storage disease characterized by the deposition of glucocerebroside in cells of the macrophage monocyte system. The disorder results from the deficiency of glucocerebrosidase. Glucocerebroside is a basic glycolipid component of the cell membrane, which is degraded by the enzyme glucocerebrosidase to glucose and lipid. Once the glucocerebroside is made it becomes a part of various cells and when these cells become old or damaged, they are often engulfed, or eaten, by immune cells called macrophages. Macrophages contain lysosomes, which are organelles that act as the cells’ digestive center. Inside lysosomes, large, potentially harmful substances are broken down, to be either discharged or reused by the body. One example is glucocerebroside which is broken down by the enzyme glucocerebrosidase which is a product of the GBA gene. In gaucher disease there are mutations in the GBA gene lead to a marked decrease in glucocerebrosidase activity. The consequences of this deficiency are generally attributed to the accumulation of the Glucocerebroside in macrophages, inducing their transformation into Gaucher cells. Under light microscopy, Gaucher cells are typically enlarged, with eccentric nuclei and condensed chromatin and cytoplasm with a heterogeneous “crumpled tissue paper” appearance. This feature is related to the presence of Glucocerebroside aggregates in characteristic twisted, fibrillar arrangements that can be visualized using electron microscopy. Gaucher cells mainly infiltrate bone marrow, the spleen, and liver, but they also infiltrate other organs. While the reason is unclear, Gaucher cells and other nearby macrophages secrete damaging lysosomal enzymes and inflammatory signals into the surrounding area. This causes an immune response and production of scar tissue, resulting in many characteristic signs and symptoms. There are a few subtypes of Gaucher disease. The most common, accounting for 99% of cases, is called type I, or the chronic nonneuronopathic form. In this type, storage of glucocerebrosides is limited to the mononuclear phagocytes throughout the body without involving the brain. Splenic and skeletal involvements dominate this pattern of the disease. Individuals with this disorder have reduced but detectable levels of glucocerebrosidase activity. In type 1, some individuals are asymptomatic, but when there are signs and symptoms they can be due to bone marrow fibrosis, which causes reduced production of red blood cells, resulting in anemia and associated fatigue. Rarely, white blood cells are also affected, causing leukopenia. There can also be bone infarctions, caused by reduced blood flow to part of a bone, can lead to a painful “bone crisis”, and result in physical deformity and avascular necrosis, or death of bone tissue. Osteoporosis, or low bone density, is another manifestation. Both the liver and spleen can become enlarged, and when platelets are sequestered, or trapped, within the enlarged spleen, this can cause thrombocytopenia, or low platelets in the blood. This can lead to bleeding and easy bruising. Type 2, or acute neuronopathic Gaucher disease, is the infantile acute cerebral pattern. In addition to the symptoms associated with type 1, there’s also loss of motor skills, decreased muscle tone, muscle spasms, and trouble swallowing.This can progress to severe breathing and feeding difficulties, and death within the first few years of life. And type 3, is intermediate between types 1 and 2. These patients have the systemic involvement characteristic of type 1 but have progressive central nervous system disease that usually begins in adolescence or early adulthood. Diagnosis of Gaucher disease relies on clinical presentation, and measuring glucocerebrosidase enzyme activity with the beta-glucocerebrosidase leukocyte blood test. Genetic testing can be done to look for mutations in the GBA gene.
Comments